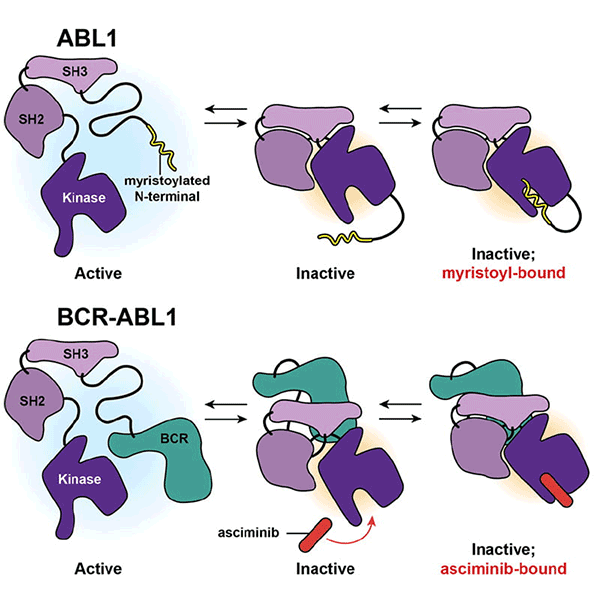
Asciminib is the first inhibitor in clinical development that binds the BCR-ABL1 myristoyl site, an allosteric site distant from the ATP site, to enforce an autoinhibited, inactive conformation. We established that asciminib, like ponatinib, is not effective against T315Iinclusive compound mutants, yet combining ponatinib (but not nilotinib or dasatinib) with asciminib is extremely effective at inhibiting many T315I-inclusive compound mutant forms of BCR-ABL1. This discovery provides the basis for a novel therapeutic strategy to address an entirely unmetmedical need and is the foundation of this proposal.
FDA approves asciminib for Philadelphia chromosome-positive chronic myeloid leukemia
In Aim 1, we will use computational, biophysical and crystallographic methods to decipher how ponatinib re-sensitizes compound mutant BCR-ABL1 to asciminib. We will test the combination in relevant mouse models and in primary leukemia samples.
In Aim 2 A, we will develop a therapeutic strategy for clinically resistant BCR-ABL1 compound mutants that are not inhibited by the combination of ponatinib with asciminib. Instead, we will target these mutants for proteasomal degradation using an asciminib proteolysis targeting chimera (PROTAC) strategy. Unlike TKIs, PROTACs are effective even upon transient or weak binding. We will test the hypothesis that ponatinib-induced stabilization of the myristoyl site is the initiating event that allows subsequent binding of an asciminib-PROTAC and proteasomal degradation of compoundmutant BCR-ABL1.
In Aim 2 B, we will develop a ponatinib-PROTAC strategy for compound mutants carrying a myristoyl site resistance mutation. Our work will provide a rationale for clinical evaluation of ponatinib combined with asciminib as a therapy for currently untreatable BCR-ABL1 compound mutant leukemia. Compound mutations are also a major cause of resistance in acute myeloid leukemia, melanoma, and lung cancer, and our study will provide a blueprint for treating thesemalignancies.
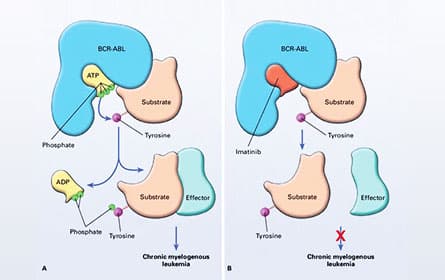 Mechanism of Action of BCR-ABL and of Its Inhibation by Imatinib.
Mechanism of Action of BCR-ABL and of Its Inhibation by Imatinib.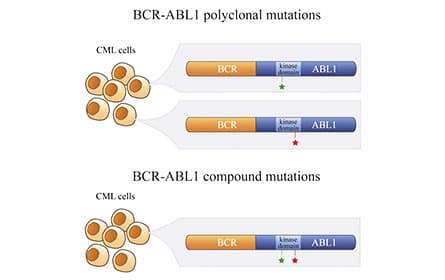 Polyclonal versus compound mutations in a subset of patients who develop clinical resistance to ABL1 TKIs.
Polyclonal versus compound mutations in a subset of patients who develop clinical resistance to ABL1 TKIs.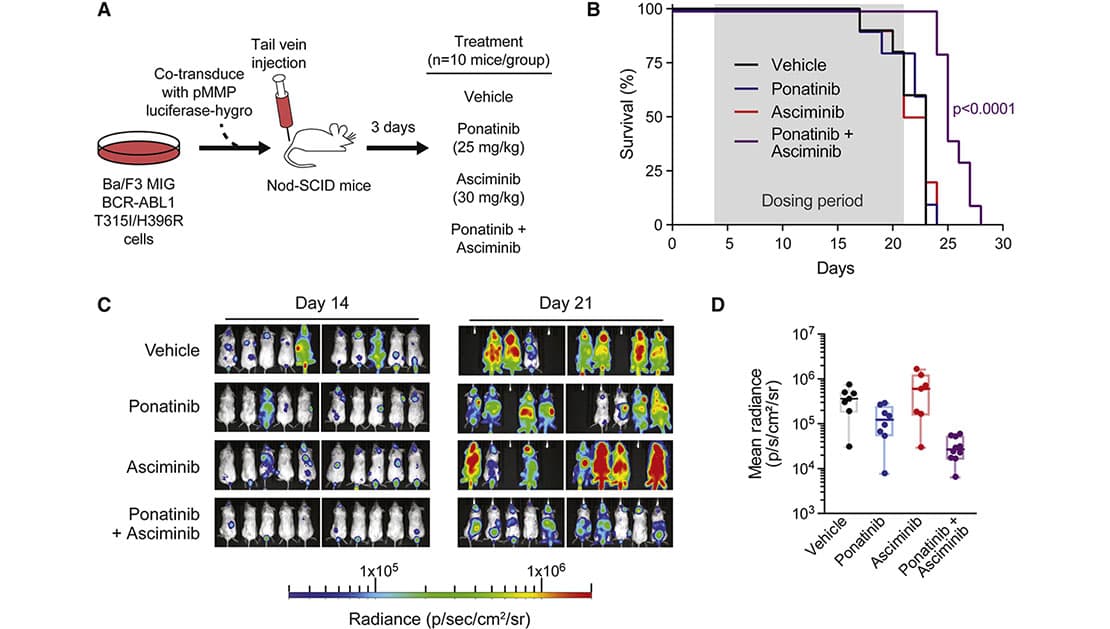 Combined Treatment with Asciminib and Ponatinib In Vivo Prolongs Survival and Inhibits T315I-Inclusive Compound Mutant Tumor Growth in a Xenograft Mouse Model
Combined Treatment with Asciminib and Ponatinib In Vivo Prolongs Survival and Inhibits T315I-Inclusive Compound Mutant Tumor Growth in a Xenograft Mouse Model